Introduction
The use of relative abundance data from next generation sequencing (NGS) can lead to misinterpretations of microbial community structures, as the increase of one taxon leads to the concurrent decrease of the other(s) in compositional data. . Since the changes of components are mutually dependent, high false discovery rates occur when compositional data are analyzed using traditional statistical methods. Although different DNA- and cell-based methods as well as statistical approaches have been developed to overcome the compositionality problem, and the biological relevance of absolute bacterial abundances has been demonstrated, the human microbiome research has not yet adopted these methods, likely due to feasibility issues. Here, we describe how quantitative PCR (qPCR) done in parallel to NGS library preparation provides an accurate estimation of absolute taxon abundances from NGS data and hence provides an attainable solution to compositionality in high-throughput microbiome analyses. The advantages and potential challenges of the method are also discussed.
Method
۱-Bacterial DNA extraction
Bacterial DNA will be extracted from fecal samples using a modified version of repeated bead beating that efficiently extracts bacterial DNA from both Gram-positive and -negative bacteria.
۲-۱۶S rRNA gene sequencing
۳-Sequencing data processing and analysis
The preprocessing will be done in the R package mare, utilizing USERACH for quality filtering, chimera removal, and taxonomic annotation. Only the high-quality forward reads should be used.
۴-Quantitative PCR
Quantification of total bacteria, specific taxa and butyrate production capacity should be carried out by qPCR.
۵-Calculation of absolute abundance and copy-number correction
The sequencing reads assigned to different taxa in each sample will be divided by the total number of reads for the sample to obtain relative abundances of the taxa in each sample. The relative abundances obtained based on the sequencing reads will be translated into total abundances by multiplying the relative abundance of each taxon by the total bacterial abundance in the sample. These figures will be further corrected for 16S rRNA gene copy-number variation by dividing the abundance of a taxon by the number of 16S copies in its genome. For the copy-number correction, the 16S copy number database rrnDB can be used.
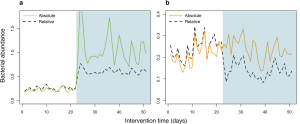
Conclusion
Importantly, qPCR-based quantitative microbiome profiling enjoys the following conceptual and practical benefits over other approaches:
۱-Cost-effectiveness and feasibility: qPCR is cost-effective and accessible as the laboratory settings, machinery and reagents are similar to those needed for preparing the NGS libraries. The same DNA extract serves as the starting material both for qPCR and NGS, making qPCR done in 96- or 384-format easy to implement in the workflow for high-throughput analysis of up to thousands of microbiome samples.
۲-Simplicity: qPCR is relatively simple to perform compared to flow cytometry that requires considerable expertise for reproducible results. In fact, flow cytometric enumeration of microbial cells was initially restricted to pure cultures and still remains challenging when performed in complex matrices [32]. Also, no spikes, other exogenous controls, or complicated transformation/computation are needed in qPCR-based quantitative microbiome profiling.
۳-Comparability to NGS: Unlike flow cytometry that counts cells, qPCR and NGS both target bacterial DNA, including extracellular DNA derived from lysed bacteria. Extracellular DNA can be intrinsic or result from the differential lysis of Gram-positive and negative bacteria during the common freeze-thawing prior to fecal DNA extraction. As the 16S profiles from the gut appear very different for intracellular and extracellular DNA [33], qPCR is expected to reflect the NGS targeted community structure both quantitatively and qualitatively more closely than flow cytometry
۴-Applicability: qPCR-based quantitative microbiome profiling is applicable also for samples containing a substantial amount of host or non-bacterial DNA, in which bacterial density cannot be reliably estimated by total DNA yield [5]. Moreover, the qPCR-based method can be employed to study also non-bacterial communities where a universal marker gene is available, such as in fungi
References
Knight R, Vrbanac A, Taylor BC, Aksenov A, Callewaert C, Debelius J, et al. Best practices for analysing microbiomes. Nature reviews Microbiology. 2018; 16(7):410–۲۲. Epub 2018/05/26. https://doi.org/10. 1038/s41579-018-0029-9 PMID: 29795328.
Morton JT, Marotz C, Washburne A, Silverman J, Zaramela LS, Edlund A, et al. Establishing microbial composition measurement standards with reference frames. Nat Commun. 2019; 10(1):2719. Epub 2019/06/22. https://doi.org/10.1038/s41467-019-10656-5 PMID: 31222023; PubMed Central PMCID: PMC6586903.
Props R, Kerckhof FM, Rubbens P, De Vrieze J, Hernandez Sanabria E, Waegeman W, et al. Absolute quantification of microbial taxon abundances. The ISME journal. 2017; 11(2):584–۷. Epub 2016/09/10. https://doi.org/10.1038/ismej.2016.117 PMID: 27612291; PubMed Central PMCID: PMC5270559.
Jian C, Luukkonen P, Yki-Järvinen H, Salonen A, Korpela K. Quantitative PCR provides a simple and accessible method for quantitative microbiota profiling. PLoS One. 2020 Jan 15; 15(1):e0227285.